
Нитрил

Во органската хемија, нитрил е секое органско соединение кое има –C≡N функционална група.[1] Префиксот цијано - се користи наизменично со терминот нитрил во индустриската литература. Нитрилите се наоѓаат во многу корисни соединенија, вклучувајќи метил цијаноакрилат, кој се користи во супер лепак и нитрилна гума, полимер што содржи нитрил кој се користи во лабораториски и медицински ракавици без латекс. Нитрилната гума е исто така широко користена како автомобилски и други заптивки бидејќи е отпорна на горива и масла. Органските соединенија кои содржат повеќе нитрилни групи се познати како цијанојаглероди.
Неоргански соединенија кои содржат–C≡N групата не се нарекуваат нитрили, туку цијаниди.[2] Иако и нитрилите и цијанидите можат да се добијат од соли на цијанид, повеќето нитрили не се ни приближно толку токсични.
Структура и основни својства
[уреди | уреди извор]Геометријата N−C−C е линеарна во нитрили, што ја одразува sp хибридизацијата на тројно врзаниот јаглерод. Растојанието C−N е кратко на 1,16 Å, во согласност со тројна врска.[3] Нитрилите се поларни, како што укажуваат високите диполни моменти. Како течности, тие имаат висока релативна дозволеност, често во 30-тите.
Историја
[уреди | уреди извор]Првото соединение од хомолошкиот ред на нитрили, нитрилот на мравја киселина, цијановодород првпат било синтетизирано од CW Scheele во 1782 година [4] Во 1811 година , Џ.Л. Геј-Лисак успеал да ја подготви многу токсичната и испарлива чиста киселина.[5] Околу 1832 година бензонитрилот, нитрилот на бензоевата киселина, бил подготвен од Фридрих Волер и Јустус фон Либиг, но поради минималниот принос на синтезата не биле утврдени ниту физичките ниту хемиските својства, ниту пак била предложена структура. Во 1834 година Теофил-Жулс Пелуз го синтетизирал пропионитрилот, сугерирајќи дека етер на пропионски алкохол и цијановодородна киселина.[6] Синтезата на бензонитрил од Херман Фелинг во 1844 година со загревање на амониум бензоат бил првиот метод што дава доволно супстанција за хемиски истражувања. Фелинг ја утврдил структурата споредувајќи ги неговите резултати со веќе познатата синтеза на цијановодород со загревање на амониум формат. Тој го измислил името „нитрил“ за новооткриената супстанција, што станало име за оваа група соединенија.[7]
Синтеза
[уреди | уреди извор]Индустриски, главните методи за производство на нитрили се амоксидација и хидроцијанизација. Двата правци се зелени во смисла дека не генерираат стехиометриски количини на соли.
Амоксидација
[уреди | уреди извор]При амоксидација, јаглеводородот делумно се оксидира во присуство на амонијак. Оваа конверзија се практикува во голем обем за акрилонитрил:[8]

Во производството на акрилонитрил, спореден производ е ацетонитрил. На индустриско ниво, неколку деривати на бензонитрил, фталонитрил, како и изобутиронитрил се подготвуваат со амоксидација. Процесот се катализира од метални оксиди и се претпоставува дека продолжува преку иминот.
Хидроцијанизација
[уреди | уреди извор]Хидроцијанизацијата е индустриски метод за производство на нитрили од цијановодород и алкени. Процесот бара хомогени катализатори . Пример за хидроцијанизација е производството на адипонитрил, претходник на најлон-6,6 од 1,3-бутадиен:

Од органски халиди и цијанидни соли
[уреди | уреди извор]Две реакции на метатеза на сол се популарни за реакции во лабораториски размери. Во Колбеловата синтеза на нитрилот, алкилхалидите се подложени на нуклеофилна алифатична супституција со цијаниди од алкални метали. Арил нитрилите се подготвуваат во синтезата Розенмунд-фон Браун.
Цијанохидрини
[уреди | уреди извор]
Цијанохидрините се посебна класа на нитрили. Класично тие произлегуваат од додавање на цијаниди од алкални метали во алдехиди во реакцијата на цијанохидрин. Поради поларитетот на органскиот карбонил, оваа реакција не бара катализатор, за разлика од хидроцијанирањето на алкените. О-силил цијанохидрините се генерираат со додавање триметилсилил цијанид во присуство на катализатор (силилцијанизација). Цијанохидрините исто така се подготвуваат со трансцијанохидрински реакции кои започнуваат, на пример, со ацетон цијанохидрин како извор на HCN.[9]
Дехидрација на амиди
[уреди | уреди извор]Нитрилите може да се подготват со дехидрација на примарните амиди. Вообичаени реагенси за ова вклучуваат фосфор пентооксид (P
2O
5) [10] и тионил хлорид (SOCl
2).[11] Во поврзаната дехидрација, секундарните амиди даваат нитрили со деградација на фон Браун амид. Во овој случај, една CN врска се расцепува.

Од алдехиди и оксими
[уреди | уреди извор]Конверзијата на алдехиди во нитрили преку алдоксими е популарна лабораториска патека. Алдехидите реагираат лесно со соли на хидроксиламин, понекогаш на температури ниски како околината, за да добијат алдоксими. Тие може да се дехидрираат до нитрили со едноставно загревање,[12] иако широк спектар на реагенси може да помогнат во тоа, вклучувајќи триетиламин / сулфур диоксид, зеолити или сулфурил хлорид. Поврзаната хидроксиламин-О-сулфонска киселина реагира слично.[13]

Во специјализирани случаи може да се користи реакцијата Ван Леузен. Биокатализаторите како што е алифатичната алдоксим дехидратаза се исто така ефикасни.
Реакција на Сендмаер
[уреди | уреди извор]Ароматичните нитрили често се подготвуваат во лабораторија од анилин преку соединенија на дијазониум. Ова е реакцијата на Сендмаер. Потребни се цијаниди од преодни метали.[14]

Други методи
[уреди | уреди извор]- Комерцијален извор за групата цијаниди е диетиалуминиум цијанид Et
2AlCN кој може да се подготви од триетилалуминиум и HCN. Се користи во нуклеофилно додавање на кетоните. - цијанидните јони го олеснуваат спојувањето на дибромидите. Реакцијата на α,α'-дибромо адипичната киселина со натриум цијанид во етанол дава цијано циклобутан:[15]

- Во таканаречената реакција на Франхимонт (која била развиена од белгискиот докторант Антоан Пол Николас Франхимонт (1844-1919) во 1872 година) а-бромокарбоксилна киселина е димеризирана по хидролиза на цијаногрупата и декарбоксилација [16]

- Ароматични нитрили може да се подготват од базна хидролиза на трихлорометил арил кетимини (RC(CCl
3)=NH) во синтезата на Хоубен-Фишер [17] - Нитрилите може да се добијат од примарните амини преку оксидација. Вообичаените методи вклучуваат употреба на калиум персулфат,[18] трихлороизоцијанурична киселина,[19] или анодна електросинтеза.[20]
- α - аминокиселините формираат нитрили и јаглерод диоксид преку различни средства за оксидативна декарбоксилација.[21][22] Хенри Драјсдејл Дакин ја открил оваа оксидација во 1916 година [23]
- Од арил карбоксилни киселини
Реакции
[уреди | уреди извор]Нитрилните групи во органските соединенија можат да подлежат на различни реакции во зависност од реактантите или условите. Нитрилната група може да се хидролизира, редуцира или исфрли од молекулата како цијаниден јон.
Хидролиза
[уреди | уреди извор]Хидролизата на нитрилите RCN продолжува во различни чекори под киселински или базен третман за прво да се добијат карбоксамидиRC(=O)NH
2 а потоа карбоксилни киселини RCOOH. Хидролизата на нитрилите до карбоксилни киселини е ефикасна. Во киселина или база, избалансираните равенки се како што следува:


Забележливо е дека овие реакции се посредувани (за разлика од катализираните) со киселина или база, бидејќи еден еквивалент на киселината или базата се троши за да се формира амониумската или карбоксилатната сол, соодветно.
Кинетичките студии покажуваат дека константата на брзина од втор ред за хидролиза на ацетонитрил во ацетамид катализирана со хидроксид-јон е 1,6 ×10−6 M −1 s −1, што е побавно од хидролизата на амидот до карбоксилат (7,4 ×10−5 M −1 s −1). Така, базниот пат на хидролиза ќе овозможи карбоксилат (или амид контаминиран со карбоксилат). Од друга страна, киселинските катализирани реакции бараат внимателна контрола на температурата и односот на реагенсите со цел да се избегне формирање на полимери, што е поттикнато од егзотермичкиот карактер на хидролизата.[24] Класичната постапка за претворање на нитрил во соодветниот примарен амид бара додавање на нитрилот во ладна концентрирана сулфурна киселина.[25] Понатамошната конверзија во карбоксилна киселина е неповолна од ниската температура и ниската концентрација на вода.

Две фамилии на ензими ја катализираат хидролизата на нитрилите. Нитрилазите ги хидролизираат нитрилите во карбоксилни киселини:

Нитрилните хидратази се металоензими кои ги хидролизираат нитрилите во амиди.
Овие ензими се користат комерцијално за производство на акриламид.
„Безводната хидратација“ на нитрилите во амиди е докажана со користење на оксим како извор на вода:[26]

Намалување
[уреди | уреди извор]Нитрилите се подложни на хидрогенизација преку различни метални катализатори. Реакцијата може да си дозволи или примарен амин (RCH
2NH
2) или терциерниот амин ((RCH
2)
3N), во зависност од условите.[27] Во конвенционалните органски редукции, нитрилот се редуцира со третман со литиум алуминиум хидрид до амин. Намалувањето на иминот проследено со хидролиза до алдехид се одвива во синтезата на Стивен алдехид, која користи олово хлорид во киселина.
Депротонација
[уреди | уреди извор]Алкилните нитрили се доволно кисели за да се подложат на депротонација на CH врската во непосредна близина на групата CN.[28][29] Потребни се силни бази, како што се литиум диизопропиламид и бутил литиум. Производот се нарекува нитрилен анјон. Овие карбаниони алкилираат широк спектар на електрофили. Клучно за исклучителната нуклеофилност е малата стерична побарувачка на единицата CN во комбинација со нејзината индуктивна стабилизација. Овие одлики ги прават нитрилите идеални за создавање на нови врски јаглерод-јаглерод во средини со стерично тешки барања.
Нуклеофили
[уреди | уреди извор]Јаглеродниот центар на нитрилот е електрофилен, па затоа е подложен на нуклеофилни реакции на додавање:
- со органоцинковно соединение во реакцијата на Блез
- со алкохоли во реакцијата на Пинер.
- со амини, на пр. реакцијата на амин саркозин со цијанамид дава креатин [30]
- Нитрилите реагираат во Фридел-Крафтс ацилација во реакцијата на Хоубен-Хоес на кетони
Разни методи и соединенија
[уреди | уреди извор]- При редуктивна децијанација нитрилната група се заменува со протон. Децијанирањето може да се постигне со растворање на редукција на метал (на пр HMPA и калиум метал во <i id="mwAWA">терц</i> -бутанол) или со соединување на нитрил во KOH.[31] Слично на тоа, α-аминонитрилите може да се децијанираат со други редукциони агенси како што е литиум алуминиум хидрид.[32]
- Нитрилите сами реагираат во присуство на база во Торповата реакција во нуклеофилен додаток
- Во органометалната хемија, познато е дека нитрилите додаваат на алкините при карбоцијанизација:[33]

Комплексирање
[уреди | уреди извор]Нитрилите се прекурсори на нитрилните комплекси на преодни метали, кои се реагенси и катализатори. Примерите вклучуваат тетракис (ацетонитрил) бакар (I) хексафлуорофосфат ([Cu(MeCN)
4]+
) и бис (бензонитрил) паладиум дихлорид (PdCl
2(PhCN)
2 ).[34]
2.jpg)
2(PhCN)
2
Деривати на нитрил
[уреди | уреди извор]Органски цијанамиди
[уреди | уреди извор]Цијанамидите се N -цијано соединенија со општа структура R1
R2
N–CN и поврзан со неорганскиот родител цијанамид.
Нитрилни оксиди
[уреди | уреди извор]Нитрилните оксиди имаат општа структура R–CNO или R–CN+
O−
и се користат во 1,3-диполарни циклододатоци.[35] :1187–1192Тие се подложени на тип 1 диотропно преуредување во изоцијанати.[35] :1700Нитрилните оксиди може да се синтетизираат со дехидрогенизација на оксимите или со дехидрација на нитроалканите. :934–936Тие можат да се користат за синтеза на изоксазоли.

Појава
[уреди | уреди извор]Нитрилите природно се појавуваат во разновидни растителни и животински извори. Над 120 природни нитрили се изолирани од копнени и морски извори. Нитрилите најчесто се среќаваат во овошните јами, особено бадемите, и за време на готвењето на културите Brassica (како зелка, бриселско зелје и карфиол), кои ослободуваат нитрили преку хидролиза. Манделонитрил, цијанохидрин произведен со внесување на бадеми или некои овошни јами, ослободува цијановодород и е одговорен за токсичноста на цијаногените гликозиди.[36]
Над 30 фармацевтски препарати кои содржат нитрил моментално се продаваат за разновидни медицински индикации со повеќе од 20 дополнителни одводи кои содржат нитрил во клиничкиот развој. Видовите на фармацевтски препарати кои содржат нитрили се различни, од вилдаглиптин, антидијабетик, до анастрозол, кој е златен стандард во лекувањето на ракот на дојката. Во многу случаи нитрилот ја имитира функционалноста присутна во супстратите за ензимите, додека во други случаи нитрилот ја зголемува растворливоста во вода или ја намалува подложноста на оксидативниот метаболизам во црниот дроб.[37] Нитрилната функционална група се наоѓа во неколку лекови.
-

-
 Структура на циталопрам, антидепресивен лек од класата на селективни инхибитори за повторно земање на серотонин (SSRI).
Структура на циталопрам, антидепресивен лек од класата на селективни инхибитори за повторно земање на серотонин (SSRI). -
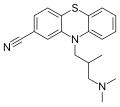 Структура на цијамемазин, антипсихотичен лек.
Структура на цијамемазин, антипсихотичен лек. -
 Структура на фадрозол, инхибитор на ароматаза за третман на рак на дојка.
Структура на фадрозол, инхибитор на ароматаза за третман на рак на дојка. -
 Структура на летрозол, орален нестероиден инхибитор на ароматаза за третман на одредени видови на рак на дојка.
Структура на летрозол, орален нестероиден инхибитор на ароматаза за третман на одредени видови на рак на дојка.





Наводи
[уреди | уреди извор]- ↑ IUPAC Gold Book nitriles
- ↑ NCBI-MeSH Nitriles
- ↑ Karakida, Ken-ichi; Fukuyama, Tsutomu; Kuchitsu, Kozo (1974). „Molecular Structures of Hydrogen Cyanide and Acetonitrile as Studied by Gas Electron Diffraction“. Bulletin of the Chemical Society of Japan. 47 (2): 299–304. doi:10.1246/bcsj.47.299.
- ↑ See:
- ↑ Gay-Lussac produced pure, liquified hydrogen cyanide in: Gay-Lussac, J (1811). „"Note sur l'acide prussique" (Note on prussic acid)“. Annales de chimie. 44: 128–133.
- ↑ J. Pelouze (1834). „Notiz über einen neuen Cyanäther“ [Note on a new cyano-ether]. Annalen der Pharmacie. 10 (3): 249. doi:10.1002/jlac.18340100302.
- ↑ Hermann Fehling (1844). „Ueber die Zersetzung des benzoësauren Ammoniaks durch die Wärme (On the decomposition of ammonium benzoate by heat)“. Annalen der Chemie und Pharmacie. 49 (1): 91–97. doi:10.1002/jlac.18440490106. On page 96, Fehling writes: "Da Laurent den von ihm entdeckten Körper schon Nitrobenzoyl genannt hat, auch schon ein Azobenzoyl existirt, so könnte man den aus benzoësaurem Ammoniak entstehenden Körper vielleicht Benzonitril nennen." (Since Laurent named the substance that was discovered by him "nitrobenzoyl" – also an "azobenzoyl" already exists – so one could name the substance that originates from ammonium benzoate perhaps "benzonitril".)
- ↑ Pollak, Peter; Romeder, Gérard; Hagedorn, Ferdinand; Gelbke, Heinz-Peter (2000), Ullmann's Encyclopedia of Industrial Chemistry, Weinheim: Wiley-VCH, doi:10.1002/14356007.a17_363
- ↑ Gregory, Robert J. H. (1999). „Cyanohydrins in Nature and the Laboratory: Biology, Preparations, and Synthetic Applications“. Chemical Reviews. 99 (12): 3649–3682. doi:10.1021/cr9902906. PMID 11849033.
- ↑ „ISOBUTYRONITRILE“. Organic Syntheses. 25: 61. 1945. doi:10.15227/orgsyn.025.0061.
- ↑ „2-ETHYLHEXANONITRILE“. Organic Syntheses. 32: 65. 1952. doi:10.15227/orgsyn.032.0065.
- ↑ Chill, Samuel T.; Mebane, Robert C. (18 September 2009). „A Facile One-Pot Conversion of Aldehydes into Nitriles“. Synthetic Communications. 39 (20): 3601–3606. doi:10.1080/00397910902788174.
- ↑ „Hydroxylamine-O-sulfonic acid: A convenient reagent for the oxidative conversion of aldehydes into nitriles“ (германски). 15 (36). 1974: 3187–3188. doi:10.1016/S0040-4039(01)91857-X. Наводот journal бара
|journal=(help) - ↑ "o-Tolunitrile and p-Tolunitrile" H. T. Clarke and R. R. Read Org. Synth. 1941, Coll. Vol. 1, 514.
- ↑ Reynold C. Fuson; Oscar R. Kreimeier; Gilbert L. Nimmo (1930). „Ring Closures in the Cyclobutane Series. II. Cyclization Of α,α′-Dibromo-Adipic Esters“. J. Am. Chem. Soc. 52 (10): 4074–4076. doi:10.1021/ja01373a046.
- ↑ A. P. N. Franchimont (1872). „Ueber die Dibenzyldicarbonsäure“ [On 2,3-diphenylsuccinic acid]. Berichte der Deutschen Chemischen Gesellschaft. 5 (2): 1048–1050. doi:10.1002/cber.187200502138.
- ↑ J. Houben, Walter Fischer (1930) "Über eine neue Methode zur Darstellung cyclischer Nitrile durch katalytischen Abbau (I. Mitteil.)," Berichte der deutschen chemischen Gesellschaft (A and B Series) 63 (9): 2464 – 2472. doi:10.1002/cber.19300630920
- ↑ Yamazaki, Shigekazu; Yamazaki, Yasuyuki (1990). „Nickel-catalyzed dehydrogenation of amines to nitriles“. Bulletin of the Chemical Society of Japan. 63 (1): 301–303. doi:10.1246/bcsj.63.301.
- ↑ Chen, Fen-Er; Kuang, Yun-Yan; Hui-Fang, Dai; Lu, Liang (2003). „A Selective and Mild Oxidation of Primary Amines to Nitriles with Trichloroisocyanuric Acid“. Synthesis. 17 (17): 2629–2631. doi:10.1055/s-2003-42431.
- ↑ Schäfer, H. J.; Feldhues, U. (1982). „Oxidation of Primary Aliphatic Amines to Nitriles at the Nickel Hydroxide Electrode“. Synthesis. 1982 (2): 145–146. doi:10.1055/s-1982-29721.
- ↑ Hiegel, Gene; Lewis, Justin; Bae, Jason (2004). „Conversion of α‐Amino Acids into Nitriles by Oxidative Decarboxylation with Trichloroisocyanuric Acid“. Synthetic Communications. 34 (19): 3449–3453. doi:10.1081/SCC-200030958.
- ↑ Hampson, N; Lee, J; MacDonald, K (1972). „The oxidation of amino compounds at anodic silver“. Electrochimica Acta. 17 (5): 921–955. doi:10.1016/0013-4686(72)90014-X.
- ↑ Dakin, Henry Drysdale (1916). „The Oxidation of Amino-Acids to Cyanides“. Biochemical Journal. 10 (2): 319–323. doi:10.1042/bj0100319. PMC 1258710. PMID 16742643.
- ↑ Kukushkin, V. Yu.; Pombeiro, A. J. L. (2005). „Metal-mediated and metal-catalyzed hydrolysis of nitriles“. Inorg. Chim. Acta. 358: 1–21. doi:10.1016/j.ica.2004.04.029.
- ↑ Abbas, Khamis A. (2008-01-01). „Substituent Effects on the Hydrolysis of p-Substituted Benzonitriles in Sulfuric Acid Solutions at (25.0± 0.1) °C“. Zeitschrift für Naturforschung A. 63 (9): 603–608. Bibcode:2008ZNatA..63..603A. doi:10.1515/zna-2008-0912. ISSN 1865-7109.
- ↑ Dahye Kang, Jinwoo Lee, Hee-Yoon Lee (2012). „Anhydrous Hydration of Nitriles to Amides: p-Carbomethoxybenzamide“. Organic Syntheses. 89: 66. doi:10.15227/orgsyn.089.0066.CS1-одржување: повеќе имиња: список на автори (link)
- ↑ Barrault, J.; Pouilloux, Y. (1997). „Catalytic Amination Reactions: Synthesis of fatty amines. Selectivity control in presence of multifunctional catalysts“. Catalysis Today. 1997 (2): 137–153. doi:10.1016/S0920-5861(97)00006-0.
- ↑ Arseniyadis, Siméon; Kyler, Keith S.; Watt, David S. (1984). „Addition and Substitution Reactions of Nitrile‐Stabilized Carbanions“. Organic Reactions. стр. 1–364. doi:10.1002/0471264180.or031.01. ISBN 9780471264187.
- ↑ Yang, Xun; Fleming, Fraser F. (2017). „C- and N-Metalated Nitriles: The Relationship between Structure and Selectivity“. Accounts of Chemical Research. 50 (10): 2556–2568. doi:10.1021/acs.accounts.7b00329. PMID 28930437.
- ↑ Smith, Andri L.; Tan, Paula (2006). „Creatine Synthesis: An Undergraduate Organic Chemistry Laboratory Experiment“. J. Chem. Educ. 83 (11): 1654. Bibcode:2006JChEd..83.1654S. doi:10.1021/ed083p1654.
- ↑ Berkoff, Charles E.; Rivard, Donald E.; Kirkpatrick, David; Ives, Jeffrey L. (1980). „The Reductive Decyanation of Nitriles by Alkali Fusion“. Synthetic Communications. 10 (12): 939–945. doi:10.1080/00397918008061855.
- ↑ The reductive decyanation reaction: chemical methods and synthetic applications Jean-Marc Mattalia, Caroline Marchi-Delapierre, Hassan Hazimeh, and Michel Chanon Arkivoc (AL-1755FR) pp. 90–118 2006 Article[мртва врска]
- ↑ Yoshiaki Nakao; Akira Yada; Shiro Ebata; Tamejiro Hiyama (2007). „A Dramatic Effect of Lewis-Acid Catalysts on Nickel-Catalyzed Carbocyanation of Alkynes“. J. Am. Chem. Soc. (Communication)
|format=бара|url=(help). 129 (9): 2428–2429. doi:10.1021/ja067364x. PMID 17295484. - ↑ Rach, S. F.; Kühn, F. E. (2009). „Nitrile Ligated Transition Metal Complexes with Weakly Coordinating Counteranions and Their Catalytic Applications“. Chemical Reviews. 109 (5): 2061–2080. doi:10.1021/cr800270h. PMID 19326858.
- ↑ 35,0 35,1 Smith, Michael B.; March, Jerry (2007). March's Advanced Organic Chemistry (6. изд.). John Wiley & Sons. ISBN 978-0-471-72091-1.
- ↑ Natural Product Reports Issue 5, 1999 Nitrile-containing natural products
- ↑ Fleming, Fraser F.; Yao, Lihua; Ravikumar, P. C.; Funk, Lee; Shook, Brian C. (November 2010). „Nitrile-containing pharmaceuticals: efficacious roles of the nitrile pharmacophore“. J Med Chem. 53 (22): 7902–17. doi:10.1021/jm100762r. PMC 2988972. PMID 20804202.
| ||||||||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||