
Генетско нарушување

Генетското нарушување — здравствен проблем предизвикан од една или повеќе абнормалности во геномот. Тоа може да биде предизвикано од мутација на еден ген (моноген) или повеќе гени (полигени) или од хромозомска абнормалност. Иако полигенските нарушувања се најчести, терминот најчесто се користи кога станува збор за нарушувања со една единствена генетска причина, било во ген или во хромозом.[1][2] Одговорната мутација може да се појави спонтано пред ембрионалниот развој (мутација de novo), или може да се наследи од двајца родители кои се носители на погрешен ген (автосомно рецесивно наследување) или од родител со нарушување (автосомно доминантно наследување). Кога генетското нарушување е наследено од еден или од двајцата родители, исто така се класифицира како наследна болест. Некои нарушувања се предизвикани од мутација на Х-хромозомот и имаат Х-поврзано наследство. Многу малку нарушувања се наследуваат на Y хромозомот или на митохондриската ДНК (поради нивната големина).[3]
Постојат повеќе од 6.000 познати генетски нарушувања,[4] и нови генетски нарушувања постојано се опишуваат во медицинската литература.[5] Повеќе од 600 генетски нарушувања се лекуваат.[6] Околу 1 од 50 луѓе се погодени од познато нарушување на еден ген, додека околу 1 од 263 се погодени од хромозомско нарушување.[7] Околу 65% од луѓето имаат некакви здравствени проблеми како резултат на вродени генетски мутации. Поради значително големиот број генетски нарушувања, приближно 1 од 21 лица е зафатено од генетско пореметување класифицирано како „ретко“ (обично дефинирано дека влијае на помалку од 1 од 2.000 луѓе). Повеќето генетски нарушувања се ретки сами по себе.[8]
Генетските нарушувања се присутни пред раѓањето, а некои генетски нарушувања предизвикуваат вродени дефекти, но вродените дефекти исто така може да бидат развојни наместо наследни. Спротивно на наследното заболување е стекната болест. Повеќето видови на рак, иако вклучуваат генетски мутации на мал дел од клетките во телото, се стекнати болести. Сепак, некои синдроми на рак, како што се мутациите <i id="mwPA">BRCA</i>, претставуваат наследни генетски нарушувања.[9]
Единечен ген[уреди | уреди извор]
| Преваленца на нарушување (приближно) | |
|---|---|
| Автосомно доминантно | |
| Семејна хиперколестеролемија | 1 од 500[11] |
| Myotonic dystrophy type 1 | 1 од 2,100[12] |
| Неврофиброматоза тип 1 | 1 од 2,500[13] |
| Сфероцитоза | 1 од 5,000 |
| Марфанов синдром | 1 од 4,000[14] |
| Хантингтонова болест | 1 од 15,000[15] |
| Автосомно рецесивно | |
| Српеста анемија | 1 од 625[16] |
| Цистична фиброза | 1 од 2,000 |
| Теј-Саксова болест | 1 од 3,000 |
| Фенилкетонурија | 1 од 12,000 |
| Автосомно рецесивно бубрежно заболување | 1 од 20,000[17] |
| Мукополисахаридоза | 1 од 25,000 |
| Волманов синдром | 1 од 40,000 |
| Болест на складирање на гликоген | 1 од 50,000 |
| Галактоземија | 1 од 57,000 |
| X-linked | |
| Дишенова мускулна дистрофија | 1 од 5,000 |
| Хемофилија | 1 од 10,000 |
| Вредностите се однесуваат за живородени бебиња | |
Едногенско нарушување (или моногено нарушување) е резултат на еден мутиран ген. Нарушувањата од еден ген може да се пренесат на следните генерации на неколку начини. Геномското втиснување и еднородителната дисомија, сепак, може да влијаат на моделите на наследување. Поделбите помеѓу рецесивните и доминантните типови не се „тешки и брзи“, иако поделбите меѓу автосомните и Х-поврзаните типови се. На пример, вообичаената форма на џуџест раст, ахондроплазија, обично се смета за доминантно нарушување, но децата со два гени за ахондроплазија имаат тешко и обично смртоносно скелетно нарушување, такво за кое ахондроплазиците може да се сметаат за носители. Српеста анемија исто така се смета за рецесивна состојба, но хетерозиготните носители имаат зголемена отпорност на маларија во раното детство, што може да се опише како поврзана доминантна состојба.[18] Кога двојката каде што едниот партнер или двајцата се засегнати или носители на нарушување од еден ген сака да има дете, тие може да го направат тоа преку ин витро оплодување, што овозможува да се изврши генетска дијагноза пред имплантација за да се провери дали ембрионот има генетско нарушување.[19]
Повеќето вродени метаболички нарушувања познати како вродени грешки на метаболизмот претставуваат резултат на нарушувања на еден ген. Многу такви нарушувања со еден ген може да ја намалат кондицијата на засегнатите луѓе и затоа се присутни кај населението на пониски фреквенции во споредба со она што би се очекувало врз основа на едноставни веројатни пресметки.[20]
Автосомно доминантно[уреди | уреди извор]
Само една мутирана копија на генот е неопходна за да едно лице биде погодено од автосомно доминантно нарушување. Секоја засегната личност обично има еден засегнат родител.[21] Шансата детето да го наследи мутираниот ген изнесува 50%. Автосомно доминантните состојби понекогаш имаат намалена пенетрација, кое означува дека иако е потребна само една мутирана копија, не сите поединци кои ја наследуваат таа мутација продолжуваат да ја развиваат болеста. Примери за овој тип на нарушување се Хантингтоновата болест,[21] неврофиброматоза тип 1, неврофиброматоза тип 2, Марфанов синдром, наследен неполипозен колоректален карцином, наследни мултипни егзостози (високо продорно автосомно доминантно нарушување), туберозна склероза, фон Вилебрандова болест и акутна интермитентна порфирија . Вродените нарушувања се нарекуваат и вродени аномалии.
Автосомно рецесивно[уреди | уреди извор]
Две копии од генот мора да бидат мутирани за личноста да биде зафатена од автосомно рецесивно нарушување. Засегнатото лице обично има незасегнати родители кои носат по една копија од мутираниот ген и се нарекуваат генетски носители. Секој родител со нарушен ген обично нема симптоми.[22] Две незасегнати единки кои носат по една копија од мутираниот ген имаат 25% ризик со секоја бременост да имаат дете погодено од ова нарушување. Примери за овој тип на нарушување се албинизам, недостаток на среднолачен ацил-CoA, цистична фиброза, српеста анемија, Теј-Саксова болест, Нимано-пикова болест, спинална мускулна атрофија и Робертсов синдром. Одредени други фенотипови, како што е влажниот наспроти сув церумен, исто така се одредуваат на автосомно рецесивен начин.[23][24] Некои автосомно рецесивни нарушувања се вообичаени затоа што, во минатото, носењето на еден од неисправните гени доведувало до мала заштита од заразна болест или токсин како туберкулоза или маларија.[25] Таквите нарушувања вклучуваат цистична фиброза,[26] српеста анемија,[27] фенилкетонурија [28] и таласемија.[29]
-
 Наследните нарушувања во ензимите генерално се наследуваат на автосомно, бидејќи има повеќе не-Х хромозоми отколку Х-хромозоми, и рецесивен начин бидејќи ензимите од незасегнатите гени генерално се доволни за да се спречат симптомите кај носителите.
Наследните нарушувања во ензимите генерално се наследуваат на автосомно, бидејќи има повеќе не-Х хромозоми отколку Х-хромозоми, и рецесивен начин бидејќи ензимите од незасегнатите гени генерално се доволни за да се спречат симптомите кај носителите. -
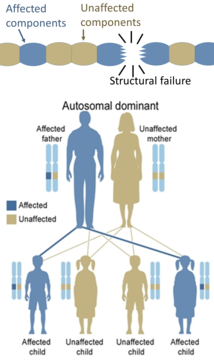 Од друга страна, наследни нарушувања во структурните протеини (како што се osteogenesis imperfecta, Марфанов синдром и многу Елерсов-Данлосови синдроми) се генерално автосомно доминантни, бидејќи доволно е што некои компоненти се неисправни за да се направи нефункционална целата структура. Ова е доминантно-негативен процес, каде што мутираниот генски производ негативно влијае на немутираниот генски производ во истата клетка.
Од друга страна, наследни нарушувања во структурните протеини (како што се osteogenesis imperfecta, Марфанов синдром и многу Елерсов-Данлосови синдроми) се генерално автосомно доминантни, бидејќи доволно е што некои компоненти се неисправни за да се направи нефункционална целата структура. Ова е доминантно-негативен процес, каде што мутираниот генски производ негативно влијае на немутираниот генски производ во истата клетка.


Х-поврзано доминантно наследување[уреди | уреди извор]

Х-поврзаните доминантни нарушувања се предизвикани од мутации во гените на Х-хромозомот. Единствено неколку нарушувања ја имаат оваа наследна шема, при што главен пример е хипофосфатемичен рахитис поврзан со Х. И мажите и жените се засегнати од овие нарушувања, при што машките обично се посериозно погодени од женките. Некои X-поврзани доминантни состојби, како што се Ретововиот синдром, инконтиненција пигменти тип 2 и Ајкардиевиот синдром, обично се фатални кај мажите, или во матката или кратко време по раѓањето, и затоа претежно се гледаат кај женките. Исклучок од ова откритие се исклучително ретки случаи во кои момчињата со Клинефелтеров синдром (44+xxy), исто така, наследуваат доминантна состојба поврзана со Х и покажуваат симптоми послични на оние на жените во однос на тежината на болеста. Шансата за пренесување на доминантно нарушување поврзано со Х се разликува помеѓу мажите и жените. Синовите на маж со доминантно нарушување поврзано со Х нема да бидат засегнати (бидејќи го добиваат хромозомот Y на нивниот татко), но сите ќерки ќе ја наследат состојбата. Жената со доминантно нарушување поврзано со Х има 50% шанси да има зафатен фетус со секоја бременост, иако во случаи како што е инконтиненција пигменти, само женските потомци се генерално остварливи.
Х-поврзано рецесивно наследување[уреди | уреди извор]
Х-поврзано рецесивно наследување е исто така предизвикано од мутации во гените на Х-хромозомот. Мажите се многу почесто погодени од жените, бидејќи тие поседуваат единствено еден Х хромозом неопходен за да се појави состојбата. Шансите за пренесување на нарушувањето се разликуваат помеѓу мажите и жените. Синовите на мажот со Х-поврзано рецесивно наследување нема да бидат засегнати (бидејќи го добиваат хромозомот Y на нивниот татко), но неговите ќерки ќе бидат носители на една копија од мутираниот ген. Жената која е носител на Х-поврзано рецесивно наследување (X R X r ) има 50% шанси да има синови кои се засегнати и 50% шанси да има ќерки кои се носители на една копија од мутираниот ген. Рецесивните состојби поврзани со Х вклучуваат сериозни болести хемофилија А, дишенова мускулна дистрофија и Леш-Ниханов синдром, како и вообичаени и помалку сериозни состојби како што се машката ќелавост и црвено-зелено слепило за бои. Х-поврзано рецесивно наследување понекогаш може да се манифестира кај женките поради искривена Х-инактивација или моносомија Х (Тарнеров синдром).
Y-поврзано рецесивно наследување[уреди | уреди извор]
Нарушувањата поврзани со Y се предизвикани од мутации на Y хромозомот. Овие состојби може да се пренесат само од хетерогаметичниот пол (на пр. мажјаци) на потомци од ист пол. Поедноставно, ова значи дека нарушувањата поврзани со Y кај луѓето може да се пренесат само од мажите на нивните синови; женките никогаш не можат да бидат засегнати бидејќи немаат Y-алозоми.
Нарушувањата поврзани со Y се исклучително ретки, но најпознатите примери обично предизвикуваат неплодност. Репродукцијата во такви услови е можна само преку заобиколување на неплодноста со медицинска интервенција.
Митохондриско наследување[уреди | уреди извор]
Овој тип на наследување, познато и како мајчинско наследство, е најретко и се однесува на 13-те гени кодирани од митохондриската ДНК. Бидејќи единствено јајце клетките придонесуваат со митохондриите во развојот на ембрионот, само мајките (кои се засегнати) можат да ги пренесат состојбите на митохондриската ДНК на своите деца. Пример за овој тип на нарушување е Леберовата наследна оптичка невропатија.
Важно е да се нагласи дека огромното мнозинство на митохондриските болести (особено кога симптомите се развиваат во раниот живот) се всушност предизвикани од нарушување на нуклеарниот ген, бидејќи митохондриите се главно развиени од не-митохондриската ДНК. Овие болести најчесто следат по автосомно рецесивно наследување.[30]
Мултифакторско нарушување[уреди | уреди извор]
Генетските нарушувања, исто така, може да бидат сложени, мултифакторни или полигенски, кое означува дека најверојатно се поврзани со ефектите на повеќе гени во комбинација со животниот стил и факторите на животната средина. Мултифакторни нарушувања вклучуваат срцеви заболувања и шеќерни болести. Иако сложените нарушувања често се групираат во семејствата, тие немаат јасен модел на наследување. Ова го отежнува одредувањето на ризикот на лицето да ги наследи или пренесе овие нарушувања. Комплексните нарушувања исто така се тешки за проучување и лекување бидејќи сè уште не се идентификувани специфичните фактори кои предизвикуваат повеќето од овие нарушувања. Проучувањата кои имаат за цел да ја идентификуваат причината за сложените нарушувања може да користат неколку методолошки пристапи за одредување на асоцијациите генотип - фенотип. Еден метод, пристапот прв генотип, започнува со идентификување на генетските варијанти кај пациентите и потоа одредување на придружните клинички манифестации. Ова се спротивставува на потрадиционалниот фенотип-прв пристап и може да ги идентификува причинските фактори кои претходно биле прикриени од клиничката хетерогеност, пенетрација и експресивност.
Според педигрето, полигенските болести имаат тенденција „да се одвиваат во семејства“, но наследството не одговара на едноставни модели како кај менделовите болести. Ова не значи дека гените на крајот не можат да се откријат и проучуваат. Постои и силна еколошка компонента за многу од нив (на пример, крвен притисок). Други фактори вклучуваат:
- астма
- автоимуни болести како мултиплекс склероза
- канцери
- цилопатии
- расцеп на непцето
- дијабетес
- срцева болест
- хипертензија
- воспалителна болест на цревата
- интелектуална попреченост
- нарушување на расположението
- дебелина
- рефрактивна грешка
- неплодност
Хромозомско нарушување[уреди | уреди извор]

Хромозомското нарушување претставува недостижен, дополнителен или неправилен дел од хромозомската ДНК.[31] Може да биде од атипичен број на хромозоми или структурна абнормалност во еден или повеќе хромозоми. Пример за овие нарушувања е Трисомија 21 (најчестата форма на Даунов синдром), во која има дополнителна копија на хромозомот 21 во сите клетки.[32]
Дијагноза[уреди | уреди извор]
Поради широкиот опсег на познати наследни генски нарушувања, дијагнозата е широко разновидна и зависи од самото нарушување. Поголемиот дел од генетските нарушувања се дијагностицираат пред породувањето, при раѓање или во раното детство, но некои, како што е Хантингтоновата болест, може да избегаат од откривање додека пациентот не почне да покажува симптоми во зрелоста.[33]
Основните аспекти на генетското нарушување се заснова на наследувањето на генетскиот материјал. Преку длабоко проучување на семејната историја, можно е да се предвидат можни нарушувања кај децата кои ги насочуваат медицинските работници на специфични тестови во зависност од нарушувањето и им овозможуваат на родителите можност да се подготват за потенцијални промени во животниот стил, да ја предвидат можноста за мртво раѓање или да размислуваат за прекин на бременоста.[34] Пренаталната дијагноза може да открие присуство на карактеристични абнормалности во развојот на фетусот преку ултразвук или да открие присуство на карактеристични супстанции преку инвазивни процедури кои вклучуваат вметнување сонди или игли во матката, како што е амниоцентезата.[35]
Прогноза[уреди | уреди извор]
Не сите генетски нарушувања директно резултираат со смрт; сепак, не постојат познати лекови за генетски нарушувања. Голем број на генетски нарушувања влијаат на фазите на развој, како што е Даунов синдром, додека други резултираат со чисто физички симптоми како што е мускулната дистрофија. Други нарушувања, како што е Хантингтоновата болест, не покажуваат знаци на болест до зрелоста. За време на активното време на генетско нарушување, пациентите најмногу се потпираат на одржување или забавување на деградацијата на квалитетот на животот и одржување на автономијата на пациентот. Ова вклучува физикална терапија и управување со болката.
Третман[уреди | уреди извор]

Третманот на генетските нарушувања е постојана битка, со над 1.800 клинички испитувања за генска терапија кои се завршени, се во тек или се одобрени ширум светот.[36] И покрај ова, најголемиот дел од опциите за третман се вртат околу лекувањето на симптомите на нарушувањата во обид да се подобри квалитетот на животот на пациентот.
Генската терапија се однесува на форма на третман каде на пациентот му се воведува здрав ген. Ова треба да го ублажи нарушувањето предизвикано од неисправен ген или да ја забави прогресијата на болеста. Главна пречка претставува испораката на гени до соодветната клетка, ткиво и орган погодени од нарушувањето. Истражувачите истражувале како можат да воведат ген во потенцијално трилиони клетки кои ја носат неисправната копија. Наоѓањето одговор на ова претставува пречка помеѓу разбирањето на генетското нарушување и корекција на генетското нарушување.[37]
Епидемиологија[уреди | уреди извор]
Околу 1 од 50 луѓе се погодени од познато нарушување на еден ген, додека околу 1 од 263 се погодени од хромозомско нарушување. Околу 65% од луѓето имаат некакви здравствени проблеми како резултат на вродени генетски мутации. Поради значително големиот број на генетски нарушувања, приближно 1 од 21 лице е зафатено од генетско нарушување класифицирано како „ретко“ (обично дефинирано дека влијае на помалку од 1 од 2.000 луѓе). Повеќето генетски нарушувања се ретки сами по себе. Постојат повеќе од 6.000 познати генетски нарушувања, и нови генетски нарушувања постојано се опишуваат во медицинската литература.
Историја[уреди | уреди извор]
Најраната позната генетска состојба кај хоминидите била во фосилниот вид Paranthropus robustus, со над една третина од поединците кои покажуваат amelogenesis imperfecta.[38]
Наводи[уреди | уреди извор]
- ↑ „Genetic Disorders“. Learn.Genetics. University of Utah. Архивирано од изворникот на 2022-07-15.
- ↑ Lvovs, D.; Favorova, O.O.; Favorov, A.V. (2012). „A Polygenic Approach to the Study of Polygenic Diseases“. Acta Naturae. 4 (3): 59–71. doi:10.32607/20758251-2012-4-3-59-71. ISSN 2075-8251. PMC 3491892. PMID 23150804.
- ↑ Reference, Genetics Home. „What are the different ways in which a genetic condition can be inherited?“. Genetics Home Reference (англиски). Архивирано од изворникот на 2020-09-27. Посетено на 2020-01-14.
- ↑ „OMIM Gene Map Statistics“. www.omim.org. Архивирано од изворникот на 2020-01-28. Посетено на 2020-01-14.
- ↑ „Orphanet: About rare diseases“. orpha.net (англиски). Архивирано од изворникот на 2019-12-17. Посетено на 2020-01-14.
- ↑ Bick, David; Bick, Sarah L.; Dimmock, David P.; Fowler, Tom A.; Caulfield, Mark J.; Scott, Richard H. (March 2021). „An online compendium of treatable genetic disorders“. American Journal of Medical Genetics. Part C, Seminars in Medical Genetics. 187 (1): 48–54. doi:10.1002/ajmg.c.31874. ISSN 1552-4876. PMC 7986124 Проверете ја вредноста
|pmc=(help). PMID 33350578 Проверете ја вредноста|pmid=(help). - ↑ Kumar, Pankaj; Radhakrishnan, Jolly; Chowdhary, Maksud A.; Giampietro, Philip F. (2001-08-01). „Prevalence and Patterns of Presentation of Genetic Disorders in a Pediatric Emergency Department“. Mayo Clinic Proceedings. 76 (8): 777–783. doi:10.4065/76.8.777. ISSN 0025-6196. PMID 11499815.
- ↑ Jackson, Maria; Marks, Leah; May, Gerhard H.W.; Wilson, Joanna B. (2018-12-03). „The genetic basis of disease“. Essays in Biochemistry. 62 (5): 643–723. doi:10.1042/EBC20170053. ISSN 0071-1365. PMC 6279436. PMID 30509934.
(calculated from "1 in 17" rare disorders and "80%" of rare disorders being genetic)
- ↑ Hunt, Jay D. „An Introduction to Cancer“. Genetics and Louisiana Families. lsuhsc.edu. Архивирано од изворникот на 16 January 2020.
- ↑ „Prevalence and incidence of rare diseases“ (PDF). Архивирано од изворникот (PDF) на 2008-11-18.
- ↑ „OMIM Entry #144010 – HYPERCHOLESTEROLEMIA, FAMILIAL, 2; FCHL2“. omim.org. Архивирано од изворникот на 2021-03-09. Посетено на 2019-07-01.
- ↑ Johnson, Nicholas E; Butterfield, Russell J; Mayne, Katie; Newcomb, Tara; Imburgia, Carina; Dunn, Diane; Duval, Brett; Feldkamp, Marcia L; Weiss, Robert B (2021). „Population Based Prevalence of Myotonic Dystrophy Type 1 Using Genetic Analysis of State-wide Blood Screening Program“. Neurology. 96 (7): e1045–e1053. doi:10.1212/WNL.0000000000011425. PMC 8055332 Проверете ја вредноста
|pmc=(help). PMID 33472919 Проверете ја вредноста|pmid=(help). - ↑ „OMIM Entry #162200 – NEUROFIBROMATOSIS, TYPE I; NF1“. omim.org (англиски). Архивирано од изворникот на 2021-03-08. Посетено на 2019-07-01.
- ↑ Keane MG; Pyeritz RE (May 2008). „Medical management of Marfan syndrome“. Circulation. 117 (21): 2802–13. doi:10.1161/CIRCULATIONAHA.107.693523. PMID 18506019.
- ↑ Walker FO (2007). „Huntington's disease“. Lancet. 369 (9557): 218–28 [221]. doi:10.1016/S0140-6736(07)60111-1. PMID 17240289.
- ↑ „OMIM Entry #603903 – SICKLE CELL ANEMIA“. omim.org (англиски). Архивирано од изворникот на 2021-04-26. Посетено на 2019-07-01.
- ↑ Swanson, Kate (2021-09-07). „Autosomal recessive polycystic kidney disease“. American Journal of Obstetrics and Gynecology. Elsevier BV. 225 (5): B7–B8. doi:10.1016/j.ajog.2021.06.038. ISSN 0002-9378. PMID 34507795 Проверете ја вредноста
|pmid=(help). - ↑ Williams T. N.; Obaro S. K. (2011). „Sickle cell disease and malaria morbidity: a tale with two tails“. Trends in Parasitology. 27 (7): 315–320. doi:10.1016/j.pt.2011.02.004. PMID 21429801.
- ↑ Kuliev, Anver; Verlinsky, Yury (2005). „Preimplantation diagnosis: A realistic option for assisted reproduction and genetic practice“. Curr. Opin. Obstet. Gynecol. 17 (2): 179–83. doi:10.1097/01.gco.0000162189.76349.c5. PMID 15758612.
- ↑ „Refinement of evolutionary medicine predictions based on clinical evidence for the manifestations of Mendelian diseases“. Scientific Reports. 9 (1): 18577. December 2019. Bibcode:2019NatSR...918577S. doi:10.1038/s41598-019-54976-4. PMC 6901466. PMID 31819097.
- ↑ 21,0 21,1 Griffiths, Anthony J.F.; Wessler, Susan R.; Carroll, Sean B.; Doebley, John (2012). „2: Single-Gene Inheritance“. Introduction to Genetic Analysis (10th. изд.). New York: W.H. Freeman and Company. ISBN 978-1-4292-2943-2.
- ↑ „Inheritance Patterns for Single Gene Disorders“. learn.genetics.utah.edu. Архивирано од изворникот на 2019-07-01. Посетено на 2019-07-01.
- ↑ Wade, Nicholas (29 January 2006). „Japanese Scientists Identify Ear Wax Gene“. The New York Times. Архивирано од изворникот на 21 March 2023. Посетено на 20 February 2023.
- ↑ Yoshiura K; Kinoshita A; Ishida T; и др. (March 2006). „A SNP in the ABCC11 gene is the determinant of human earwax type“. Nat. Genet. 38 (3): 324–30. doi:10.1038/ng1733. PMID 16444273.
- ↑ Mitton, Jeffry B. (2002). „Heterozygous Advantage“. eLS. doi:10.1038/npg.els.0001760. ISBN 978-0-470-01617-6.
- ↑ „Evaluating candidate agents of selective pressure for cystic fibrosis“. Journal of the Royal Society, Interface. 4 (12): 91–8. February 2007. doi:10.1098/rsif.2006.0154. PMC 2358959. PMID 17015291.
- ↑ „Genetic control of resistance to human malaria“. Current Opinion in Immunology. 21 (5): 499–505. October 2009. doi:10.1016/j.coi.2009.04.001. PMID 19442502.
- ↑ Woolf, LI (1986). „The heterozygote advantage in phenylketonuria“. American Journal of Human Genetics. 38 (5): 773–5. PMC 1684820. PMID 3717163.
- ↑ Weatherall, D. J. (2015). „The Thalassemias: Disorders of Globin Synthesis“. Williams Hematology (9e. изд.). McGraw Hill Professional. стр. 725. ISBN 9780071833011. Архивирано од изворникот на 2023-02-20. Посетено на 2023-02-20.
- ↑ Nussbaum, Robert; McInnes, Roderick; Willard, Huntington (2007). Thompson & Thompson Genetics in Medicine. Philadelphia PA: Saunders. стр. 144, 145, 146. ISBN 9781416030805.
- ↑ „Genetic Disorders: What Are They, Types, Symptoms & Causes“. Cleveland Clinic (англиски). Архивирано од изворникот на 2023-11-01. Посетено на 2023-11-01.
- ↑ CDC (2023-10-10). „Facts about Down Syndrome | CDC“. Centers for Disease Control and Prevention (англиски). Архивирано од изворникот на 2017-07-28. Посетено на 2023-11-01.
- ↑ Wyant, Kara J.; Ridder, Andrew J.; Dayalu, Praveen (21 March 2017). „Huntington's Disease — Update on Treatments“. Current Neurology and Neuroscience Reports. 17 (4). doi:10.1007/s11910-017-0739-9.
- ↑ Milunsky, Aubrey; Milunsky, Jeff M. (2021). „Genetic Counseling: Preconception, Prenatal, and Perinatal“. Genetic Disorders and the Fetus. стр. 1–101. doi:10.1002/9781119676980.ch1. ISBN 978-1-119-67698-0.
- ↑ „Diagnostic Tests – Amniocentesis“. Harvard Medical School. Архивирано од изворникот на 2008-05-16. Посетено на 2008-07-15.
- ↑ Ginn, Samantha L.; Alexander, Ian E.; Edelstein, Michael L.; Abedi, Mohammad R.; Wixon, Jo (February 2013). „Gene therapy clinical trials worldwide to 2012 – an update“. The Journal of Gene Medicine. 15 (2): 65–77. doi:10.1002/jgm.2698. PMID 23355455.
- ↑ Verma, I. M. (22 August 2013). „Gene Therapy That Works“. Science. 341 (6148): 853–855. Bibcode:2013Sci...341..853V. doi:10.1126/science.1242551. PMID 23970689.
- ↑ Towle, Ian; Irish, Joel D. (April 2019). „A probable genetic origin for pitting enamel hypoplasia on the molars of Paranthropus robustus“ (PDF). Journal of Human Evolution. 129: 54–61. doi:10.1016/j.jhevol.2019.01.002. PMID 30904040. Архивирано од изворникот (PDF) на 2023-06-04. Посетено на 2023-02-20.
Надворешни врски[уреди | уреди извор]
- Public Health Genomics at CDC
- OMIM — Online Mendelian Inheritance in Man, a catalog of human genes and genetic disorders
- Genetic and Rare Diseases Information Center (GARD) Office of Rare Diseases (ORD), National Institutes of Health (NIH)
- CDC’s National Center on Birth Defects and Developmental Disabilities
- Genetic Disease Information from the Human Genome Project
- Global Genes Project, Genetic and Rare Diseases Organization
- List of Genetic Disorders - Genome.gov